|
Heslop-Harrison Group Homepage Science Bovid (cow) diversity People Methods/techniques In situ Hybridization Molecular methods- lab protocols Technology development Courses and Science Pictures Journal editing Policy Information/Publications Jack Heslop-Harrison Links Site information
|
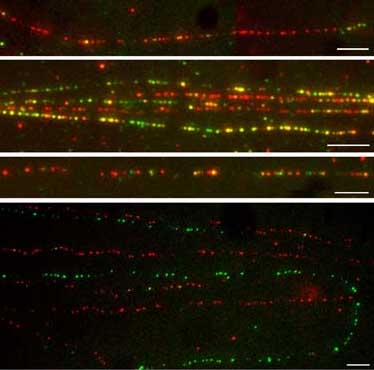
Pat Heslop-Harrison and Trude Schwarzacher (Techniques based on Michalet, Bensimon et al. 1997 and Jiming Jiang, Madison, Wisconsin) Reagents - Methods - Troubleshooting/key points - Alternatives - References Since 2006, we have not used the pulling method so much. Aaron Bensimon now has a company Genomic Vision (and www.igmm.cnrs.fr which offers both the equipment and treated slides, although we have not tried them. If I were to build another system for pulling, I would use the Lego Technic system which has lots of gears and operates very slowly and smoothly, as well as being very cheap (and more fun) compared to buying motors, gears, supports from lab. supplies companies. Getting the slides just right to hold the DNA is important. After trying various slide sources and our own coated ones, recently we have be using poly-L-lysine slides from Muto Pure Chemicals, Tokyo, Japan. These we get/bring from Japanese collaborators, and I do not think they are available outside Japan but any lab. there can obtain them. This is no guarantee that another batch bought two years later would still be good in DNA fibre fiber spreading applications.. Pictures of our current type of results are in papers at http://www.le.ac.uk/biology/phh4/openpubs/Dros_junctions_kuhn.pdf (excerpt shown above left) from Heredity and Chromosome Research http://www.le.ac.uk/biology/phh4/HHPubs/buzzsat.pdf (intranet only) (or doi: 10.1007/s10577-007-1195-1 ). The latter gives the protocol we are using now in good detail (based on one from Jiming Jiang in Wisconsin), and we have continued to use this most recently. This paper gives the protocol for Drosophila, but the same approach has worked for plants. For DNA fibre spreading, high-molecular-weight DNA for in situ hybridization was prepared from fresh Drosophila adults or flies stored at -80C. The protocol used was adapted from Schwarzacher & Heslop-Harrison (2000) and from an online protocol provided by Jiming Jiang formerly at http://www.hort.wisc.edu/jjiang/nuclear_fiber_fish_1.htm (not available 9/2011).. Approximately 10 to 20 flies were homogenized with a pestle in a microcentrifuge tube containing 500 ul Nuclear Isolation Buffer (NIB: 10 mM Tris-HCl pH 9.5, 10 mM EDTA, 100 mM KCl, 0.5 M sucrose, 4.0 mM spermidine, 1.0 mM spermine, 0.1% mercaptoethanol) and subsequently filtered twice through nylon mesh filters. After addition of 5 ul (micro-litres) of NIB containing 10% Triton X-100 (pre-mixed, final concentration between 0.5% and 1%), the solution was centrifuged (2000 g; 10 min) and the pellet was resuspended in 100-200 ul of 1:1 NIB (without mercaptoethanol) and 100% glycerol. An aliquot of the nuclear suspension (c 10 ul) was transferred to a microcentrifuge tube containing 100 ul NIB, followed by centrifugation (1200 g; 5 min). The pellet of nuclei was subsequently resuspended in 20 ul PBS (10 mM sodium phosphate, pH 7.0, 140 mM NaCl). In one end of a poly-L-lysine slide (Muto Pure Chemicals, Tokyo, Japan), 2 ul of nuclear suspension was added, air-dried for 5Y10 min (but avoiding overdrying) and 8 ul of STE lysis buffer (0.5% w/v SDS, 100 mM Tris-HCl, pH 7.0 and 50 mM EDTA) was added above the nuclei drop. The slides were incubated for 6-10 min (avoiding overdrying) at room temperature and the solution was dragged down the slide with the edge of a coverslip. The slides were air-dried at room temperature, fixed in fresh 3:1 ethanol:acetic acid, air-dried and incubated at 60-C (30 min). The in situ hybridization steps and post-hybridization washes were conducted basically as described for chromosomes, with DNA fibers counterstained with DAPI. The length of single DNA fibers was measured considering that 320 pixels (objective 100) or 180 pixels (objective 63) correspond to 10 mm, which corresponds to 29 kb of the extended DNA fiber (Schwarzacher & Heslop-Harrison 2000). Typically, two slides per species were analyzed and for each slide more than 10 pictures from different slide areas were captured. Reagents YOYO-1 DNA stain (Molecular Probes - Invitrogen): dilute in water to 5 nM. MES (2-[N-Morpholino]ethanesulfonic acid; Sigma M-8250): stock solution of 0.5mM, adjusted to pH 5.5 with KOH. Glue: cyanoacrylate (‘superglue’) Materials Cover-slips coated with silane solution (10% (v/v) 3-aminopropyltriethoxysilane (APES) in acetone) 5 minutes. Rinse three times with deionized water (washing step seems crucial to me.) Teflon reservoir: block of Teflon (that is, PTFE plastic,
very slippy, DNA does not stick to it) milled with a slot 4-5 mm wide,
length and depth to fit coverslip (18x18 or 22x22 mm). This can be made
easily with a drill, and the piece of Teflon costs less than USdollar
$1, from e.g. model shop or electronics supply shop (as insulation for
power transistors). Pulling device: another trip to the model shop! Obtain a battery-operated motor, with basic speed control and a gear box allowing speeds of a few revolutions per minute (rpm). Plastic gearboxes with adjustable gears are available (e.g. RS components in UK) for a few dollars. Clamp motor and gears to a retort stand, attach thread to output spindle, pass over the top of the retort stand and onto a clip (either a bulldog-type paper clip or reversed, self-closing forceps) holding the cover-slip in the Teflon reservoir. Extra weight may be needed to ensure smooth movement. Method
10 minutes in paraformaldehyde 2 washes of 5' in 2x SSC Alcohol dehydratation (80-100%), and air dry Add probes, denature 8 minutes at 85�C Hybridize Overnight at 37�C Three washes of 5 minutes in formamide 50% 2x SSC at
room temperature Three washes of 5 minutes in 2x SSC 5 minutes with 0.15g BSA in 4x SSC/0.1% Tween 20 Detection with antiDig and antiBio, one hour at 37�C Three washes of 5 minutes in 4x SSC/Tween Mounting with Anti-fade, e.g. Citifluor. Troubleshooting, key points and modifications Dissolving DNA in a drop of buffer and running down the sloping slide very slowly. Embedding DNA in agarose blocks as for PFGE, placing on microscope slide, meltiing and dragging with a coverslip. These will appear in more detail here; meanwhile see Practical In situ hybridization book.
|